Biology is spatial
At Spatial Genomics, we are dedicated to empowering discovery. We believe that highly multiplexed spatial analysis will advance fundamental biology, precision medicine, drug development, and diagnostics.
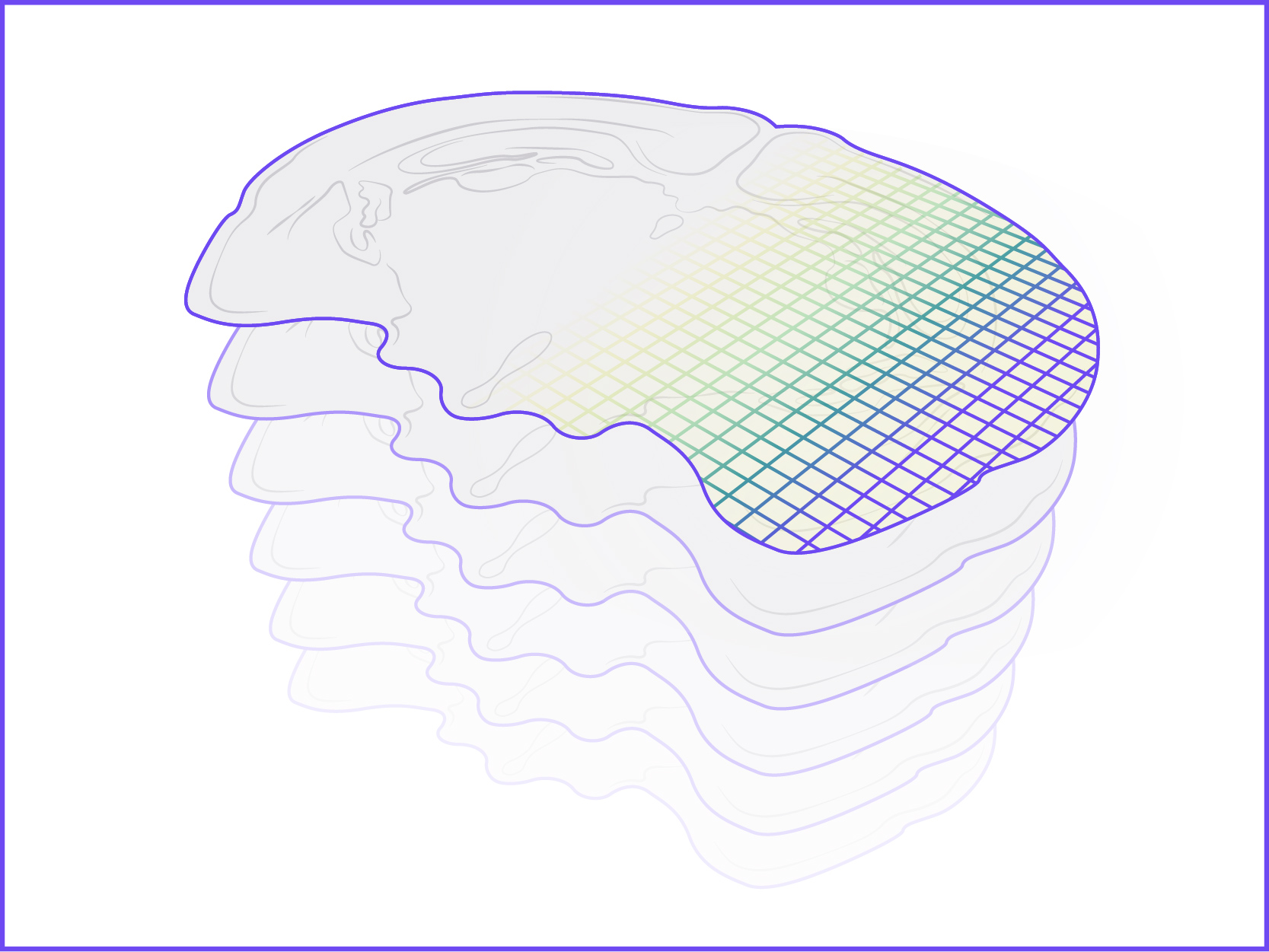
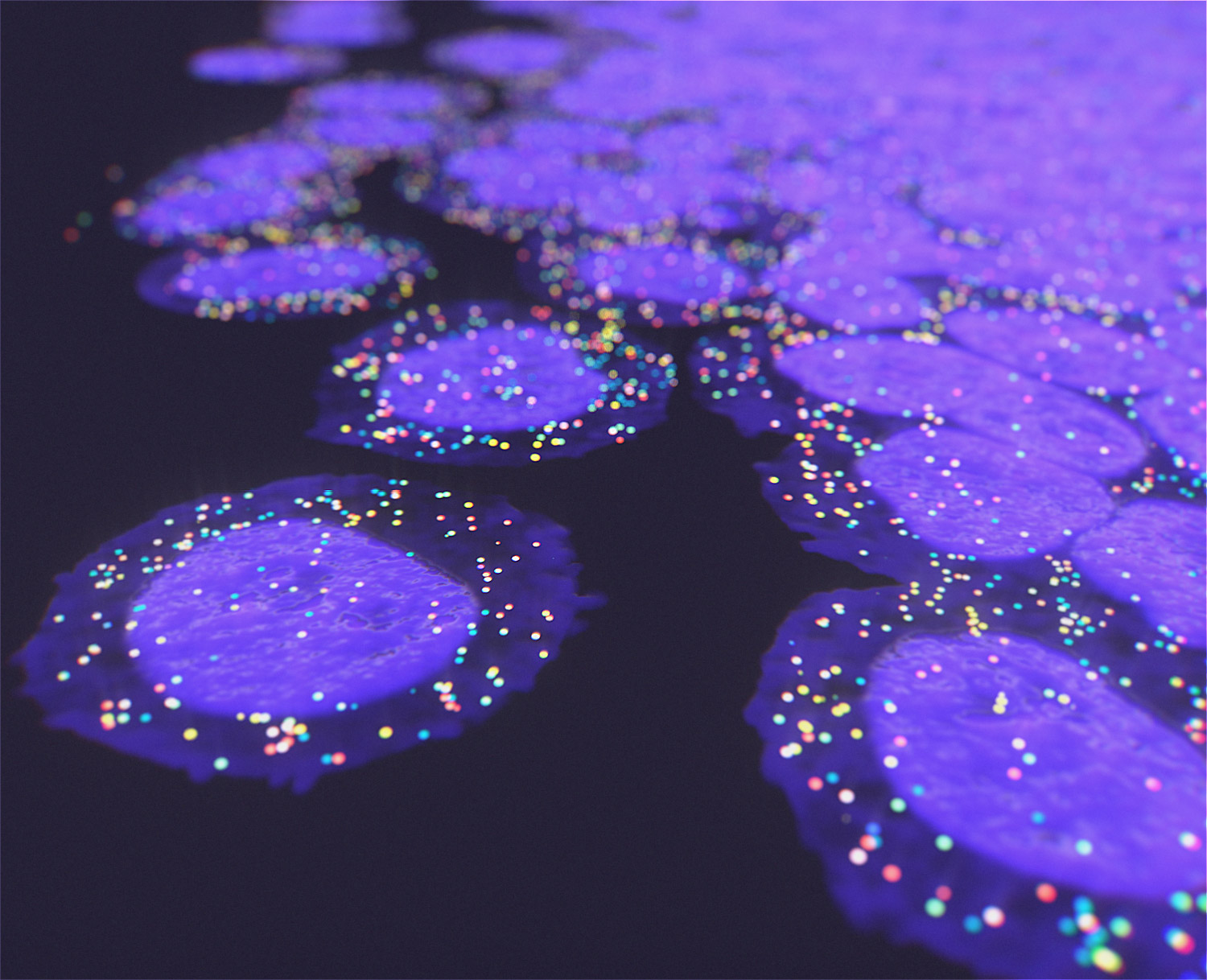
We offer the new GenePS device to enable spatial research. This sensitive confocal imager is combined with advanced fluidics to deliver excellent results. Off-the-shelf and custom gene panels are available to meet your project needs and powerful analysis software allows you to quickly review the results.
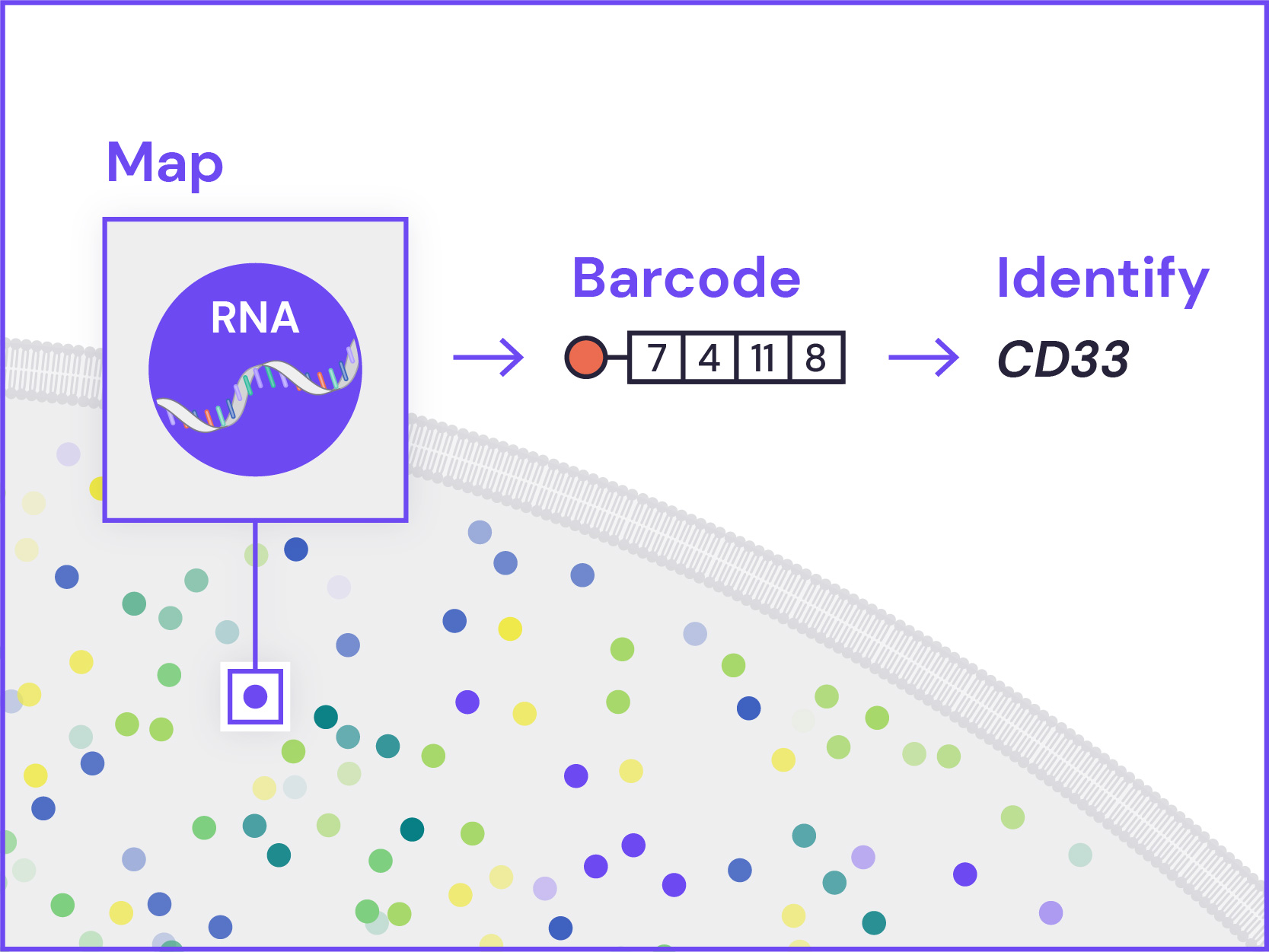
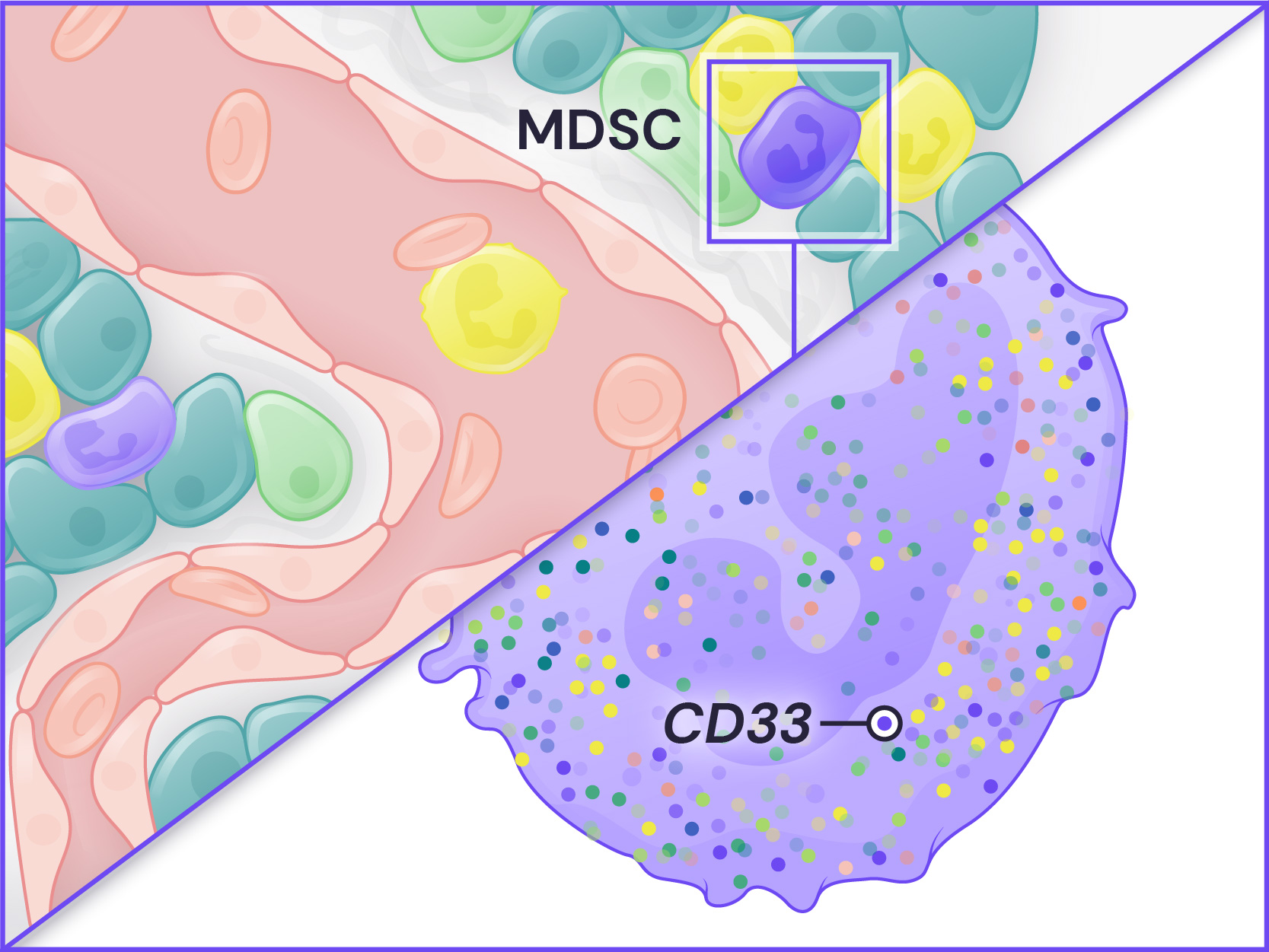
Spatial Genomics reveals biology. The GenePS can assay single-cell gene expression, protein expression, and genomic and epigenomic states within intact cells and tissues. As a result, we catalog more than cell states and are better able to assess cell neighborhoods, interactions, and influences.